
.svg.png)





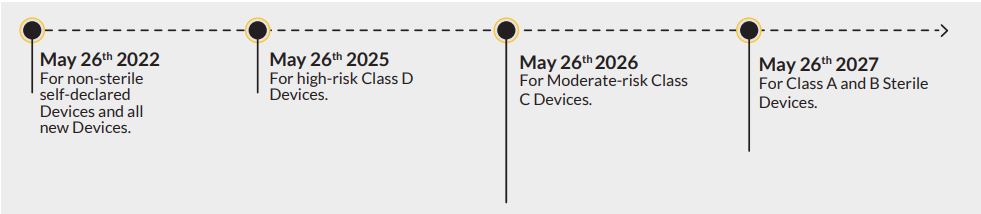
In May 2017, EU officials announced regulations impacting organizations that manufacture in vitro diagnostic devices in both Europe and the United States. In order to sell their products on the European market, organizations must achieve compliance with these new regulations by May 2022. When you consider the challenges ahead, it makes sense to start taking steps to prepare – especially since some deadlines have already come and gone.
It’s complicated.
Understanding the distinct challenges that will impact product reviews and approvals can be difficult. Many guidance documents are still in development, which leaves manufacturers with more questions than answers.
It’s extensive.
Conducting a gap assessment is a good first step; however, manufacturers will need to manage more than gaps alone. This means evaluating not only your products, but also your processes. The new focus of these regulations is on product and process, with a focus on risk-assessment.
It’s risky.
If issues are not addressed early enough, you run the risk of failing to pass audits or conformity assessments, which could result in your products being restricted from market distribution. In addition, the Notified Bodies will have limited capacity, which will likely cause additional delays due to backlog.
It’s specific.
One section that requires careful attention is the “management by exception and memorandum.” This specifies that manufacturers must have supporting data and objective evidence. Do you have all your documentation in place for this change?
